|
Function runLabPolymer.m
Simple Polymer simulatorWe set up a simple polymer simulator with a cartesian grid and physical boundary conditons such as given influx in input cells and given pressure, saturation and concentration on output faces. function [G, states] = runLabPolymer()
Add mrst modules which are used mrstModule add ad-fi deckformat mrst-gui ad-props Input data are read from a file. The format is the same as Eclipse. Values are store in the deck structure. The input file contains the fluid and polymer properties. current_dir = fileparts(mfilename('fullpath')); fn = fullfile(current_dir, 'LABPOLYMER.DATA'); deck = readEclipseDeck(fn); The deck variables are converted to SI units. MRST uses exclusively SI units in the computation. Conversion tools are available. deck = convertDeckUnits(deck); Generate grid. We set up two cases: 1D or 2D. The changes in the remaining of the code are minimal. They appear only in the file setupControls.m where the boundary conditions are set. sim_case = '1D'; % '1D' or '2D' switch sim_case case '1D' nx = 1000; ny = 1; nz = 1; xlength = 10; ylength = 1; zlength = 1; case '2D' nx = 100; ny = 100; nz = 1; xlength = 10; ylength = 10; zlength = 1; end The function ¦cartGrid¦ generates a MRST unstructured grid. The function computeGeometry computes geometrical properties such as cell volumes, faces areas... G = cartGrid([nx ny nz], [xlength, ylength, zlength]); G = computeGeometry(G); Setup rock structure containing the rock properties. rock.perm = repmat(100*milli*darcy, [G.cells.num, 1]); rock.poro = repmat(0.3, [G.cells.num, 1]); Setup fluid structure containing the fluid and polymer properties form the deck structure. fluid = initDeckADIFluid(deck); We set off gravity. gravity off
Setup adsorption function. There are two options: with or without desorption. fluid.effads = @(c, cmax) effads(c, cmax, fluid); Setup the inputs. They are given as boundary conditions and sources Injection will done in given cells. bc.injection.rate = 0.1/day; bc.injection.s = 1; bc.injection.c = 1; pressure, saturation and polymer concentration are imposed on given faces bc.dirichlet.pressure = 100*barsa; bc.dirichlet.s = 0; bc.dirichlet.c = 0; The function setupControls.m set up the bc structure. bc = setupControls(G, bc, sim_case); The function setupSystem.m sets up the system. In particular, it defines the discrete differential operators. system = setupSystem(G, rock, bc); system.fluid = fluid; system.rock = rock; system.nonlinear.tol = 1e-4; system.nonlinear.maxIterations = 30; system.nonlinear.relaxRelTol = 0.2; system.cellwise = 1:3; The transport part can be either solved explicitly or implicitly. From the implementation point of view, it corresponds to one line of change in the code, see equationOWPolymer.m system.implicit_transport = false; Set up the initial state with constant pressure, saturation and polymer concentration. nc = G.cells.num; init_state.pressure = 1*atm*ones(nc,1); init_state.s = ones(nc, 1)*[0.2, 0.8]; init_state.c = zeros(G.cells.num, 1); init_state.cmax = zeros(G.cells.num, 1); Compute and store the mobilities for the Dirichlet values [bc.mobW, bc.mobO, bc.mobP] = computeMobilities(bc.dirichlet.pressure, ... bc.dirichlet.s , ... bc.dirichlet.c , ... bc.dirichlet.c , ... fluid); Set up time stepping parameters. We use a fixed time step. total_time = 2*day; dt = 0.001*day; steps = dt*ones(floor(total_time/dt), 1); t = cumsum(steps); We start the time step iterations. Each iteration is solved fully implicitly. Time step iterations. state0 = init_state;
nsteps = numel(steps);
states = cell(nsteps, 1);
for tstep = 1 : nsteps
dt = steps(tstep); Call non-linear solver solvefi.m to compute next state [state, conv, its] = solvefi(state0, dt, bc, system);
if ~(conv)
error('Convergence failed. Try smaller time steps.')
return
else
fprintf('Step %d completed in %d Newton iterations\n', tstep, its);
end
states{tstep} = state;
state0 = state;
end
plotState(state, G.cells.centroids(:, 1));
end
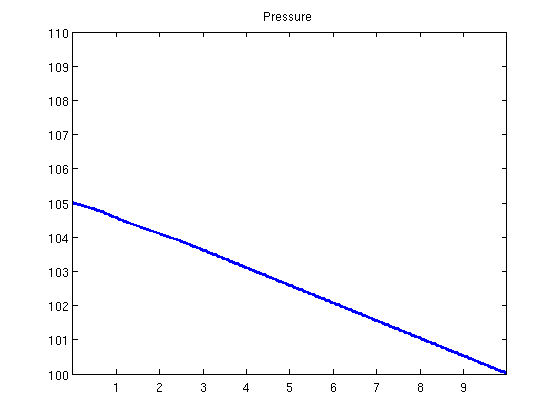
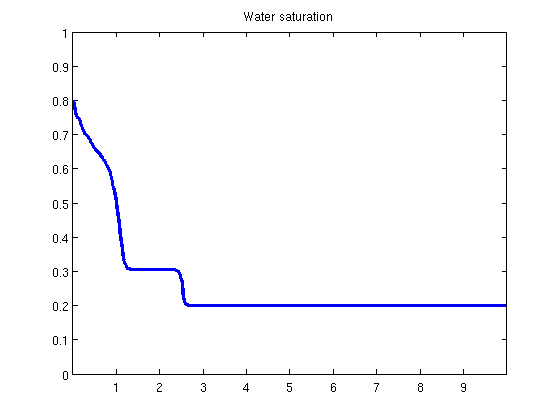
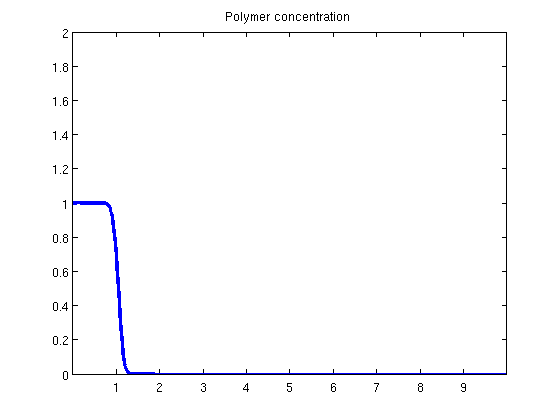
Helper functionfunction y = effads(c, cmax, f) if f.adsInx == 2 y = f.ads(max(c, cmax)); else y = f.ads(c); end end function plotState(state, xval) figure(1) plot(xval, state.pressure/barsa); axis([xval(1), xval(end), 100, 110]); title('Pressure') figure(2) plot(xval, state.s(:, 1)); axis([xval(1), xval(end), 0, 1]); title('Water saturation') figure(3) plot(xval, state.c); axis([xval(1), xval(end), 0, 2]); title('Polymer concentration') end |
||||||||