|
|

|
Theoretical tools
Molecular modeling has become a necessary tool in state-of-the-art catalysis research, and it has been prominent in the CATMAT collaboration. Reaction and activation energies may be computed, allowing for predictions of thermodynamics, kinetics, and reaction mechanisms. Modelling is most useful when it is combined with experimental methods in a close interdisciplinary approach, and this is how we apply modelling in CATMAT.
Methods:
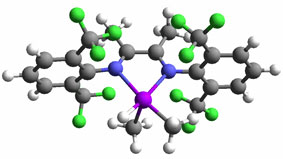
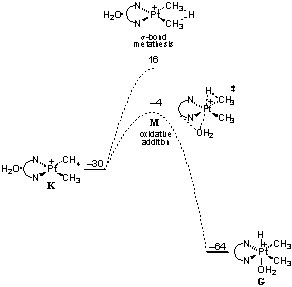
Some cases: Heterolytic activation of methane by a homogeneous platinum catalyst Methanol to hydrocarbons
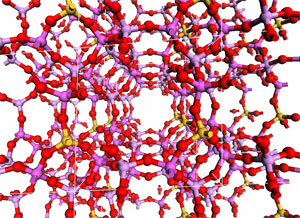
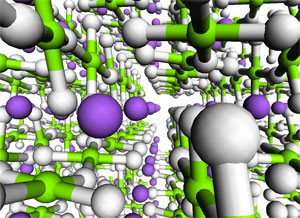
Other aspects of the mechanisms require explicit modelling of the solid. In these cases, we resort to so-called band structure models. We are in the process of investigating acid catalysis with such extended models. The picture illustrates 9 unit cells of the catalytic material SAPO-34. (See publication list) Complex hydrides as hydrogen storage materials The computational screening of different materials markedly improves the efficiency of the search for the optimal hydrogen storage medium, as experimental work may be focussed on the candidates that are predicted to have the most desirable properties.
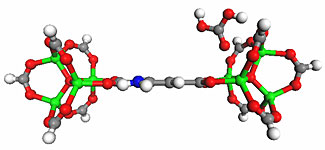
Metal Organic framework as selective sorption of CO2
See publications list for Theoretical tools |
||||||||